
Por: María Fontan Sanz
Las descripciones clínicas de la distrofia muscular de Duchenne (DMD) se han producido desde mediados de 1800. Es una enfermedad con pérdida muscular lentamente progresiva caracterizada por el desarrollo de síntomas antes de los 5 años de edad. Aunque esta enfermedad recibe el nombre del doctor francés Duchenne de Boulogne fue Edward Meryon quien hizo las primeras descripciones en 1852. Estudió biopsias musculares realizadas a 4 hermanos que presentaban un tipo de distrofia muscular y observó la presencia de alteraciones patológicas tales como la disrupción del sarcolema. Para caracterizar su curso clínico realizó un informe detallado de la vida de un paciente, quien en su infancia presentaba un retraso a nivel motor y nunca había podido saltar. A la edad de 8 años tenía dificultad para subir escaleras, a los 11 años no podía mantenerse en bipedestación y a los 14 años sus extremidades superiores se habían debilitado completamente, muriendo a los 16 años.
En 1861 Duchenne describió a un grupo de pacientes con paraplejía hipertrófica de gastrocnemios e inicialmente pensó que la causa era de origen cerebral. Pero hacia 1868, tras estudios repetidos de múltiples biopsias musculares, consideró que la enfermedad era de origen muscular y describió en una publicación los signos y síntomas de la DMD. En 1886 Gowers describe el signo clásico que lleva su nombre después de observar a varios niños con DMD (aunque el signo de Gowers no es específico de la DMD). A lo largo de estos años y a pesar de que algunos médicos del momento ya eran conscientes de la existencia de varios tipos de distrofia muscular no hubo avances significativos en el estudio de la enfermedad hasta casi mediados del S. XX.
A partir de 1930 diferentes grupos de investigadores describen el aumento de creatina kinasa (CK), tanto en pacientes con diagnóstico de la enfermedad como en algunas mujeres portadoras (elevación moderada en portadoras). En 1950 el doctor alemán Emil Becker descubre otro tipo de distrofia muy similar a la DMD. Watson y Crick proponen el modelo de doble hélice de ADN en 1953, lo cual supuso una contribución significativa al estudio de distintas enfermedades. Surgen los primeros estudios que indican el beneficio de la prednisona en la DMD. En 1980, más de 100 años después de que Duchenne describiera la enfermedad, Louis Kundel se dispone a encontrar la causa de la DMD en el Hospital Infantil de la Universidad de Harvard. Pidió a la MDA financiación para su proyecto pero no se la concedieron ya que no creyeron que fuera capaz de localizar el gen defectuoso. Kundel presentó un proyecto en el que explicaba cómo iba a localizar el gen defectuoso, identificar su secuencia, estudiar el tipo de mutación y la secuencia de aminoácidos de la proteína para la que codificaba ese gen. Kundel sospechaba que el gen causante de la enfermedad debía estar en el cromosoma X; y también sospechaba el patrón por el que se transmitía (recesivo) ya que eran los niños varones los que desarrollaban la enfermedad y sus madres eran en muchas ocasiones portadoras genéticas. Finalmente convenció a la MDA y logró la financiación.
Hacia 1986 Kundel y sus colaboradores aislan el gen DMD. Finalmente en 1987 Hoffman y Cambpell, del equipo de Kundel, identifican la proteína producto de este gen a la que llaman distrofina. Se realizaron experimentos inyectando esta proteína a conejos, los cuales generaban anticuerpos frente a ella. Tras marcar los anticuerpos con marcadores fluorescentes se vio que la distrofina estaba localizada bajo la membrana de la fibra muscular (subsarcolémica). Además comprobaron que había un déficit de distrofina y que la cuantía de este déficit estaba relacionada con la gravedad de la DMD.La DMD estaba asociada a niveles altos de CK en sangre porque al dañarse el sarcolema se libera CK, entre otras sustancias, desde el interior de la célula muscular.
Desde finales de los 90 se han realizado diversos experimentos animales con la finalidad de encontrar posibles tratamientos. La prednisona retrasa la progresión de la enfermedad y la pérdida de fuerza muscular pero no modifica la evolución fatal de la enfermedad. A partir del 2000 se estudian posibles nuevas terapias génicas como el salto del exón (exón skipping, omisión de exón) mediante la administración de oligonucleótidos antisentido, lo cual puede hacerse utilizando un virus como vector. En la DMD el gen de la distrofina puede presentar deleciones (70-80%), duplicaciones (7-11%) o mutaciones puntuales (10-30%). Los oligonucleótidos antisentido son moléculas que se ensamblan con el pre-RNA-m en el lugar de la mutación y permiten saltar u omitir el exón o exones mutados para que se continúe la pauta de lectura. El RNA-m resultante es más corto con lo que se produce una proteína más corta pero parcialmente funcional. Es decir, el salto de exón intenta restaurar la pauta de lectura mediante la supresión de uno o más exones y pretende mitigar la enfermedad convirtiendo una forma de Duchenne en una de Becker, la cual presenta menor gravedad.
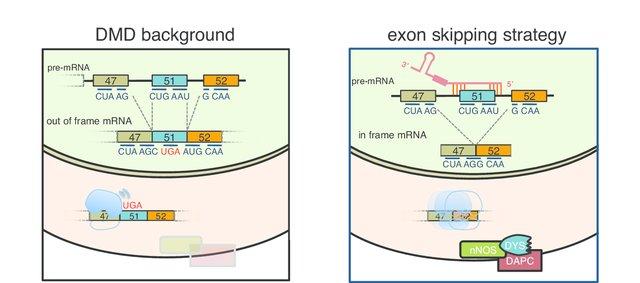
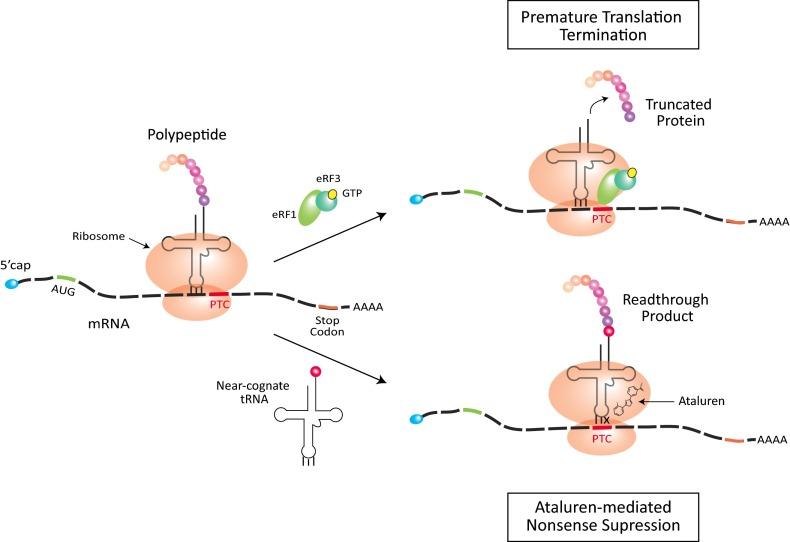
Esta terapia se ha desarrollado durante los últimos 15 años por varios grupos de investigación, sobre todo en Japón, Australia, Reino Unido y los Estados Unidos y se ha aplicado a ratones y perros con buenos resultados. El mecanismo de salto de exón 51 es el más desarrollado, seguido de los exones 44, 45, 52, 53. A partir del año 2000 se comienza a estudiar la administración de Traslarna (Atalureno), que es un compuesto químico destinado a restaurar la lectura del RNA-m en pacientes que portan una mutación sin sentido (nonsense). Las mutaciones sin sentido dan lugar a un codón de parada prematuro en el RNA-m, con lo que se va a finalizar la traducción antes de que se genere una proteína funcional. Ataluren facilita la lectura ribosómica del RNA-m que contiene el codón de parada prematuro, lo que tiene como resultado la producción de una proteína funcional.
A partir del 2005 se hacen estudios en ratones en los que se induce la síntesis de proteínas endógenas similares a la distrofina, como la utrofina. Posteriormente se han realizado estudios preliminares en voluntarios humanos. Se propone que la regulación al alza de la utrofina en el músculo de pacientes con DMD podría reducir significativamente la degeneración muscular, compensando al menos parcialmente la ausencia de distrofina. En el 2013 un equipo de investigación de la Universidad de Missouri identifica en el gen de la distrofina secciones que son esenciales para el buen funcionamiento de los tejidos musculares. En 2014 la Agencia Europea del Medicamento aprueba el primer medicamento para el tratamiento de un grupo de mutaciones específicas de la DMD (Atalureno) mientras que la FDA lo hace en el 2016 (Eteplirsen, salto de exón 51).
En 2018 La Universidad de Duke ha cultivado con éxito tejido funcional a partir de una muestra de piel, lo que permite nuevos enfoques terapéuticos para la regeneración tisular en la DMD. En la actualidad investigadores en el Hospital y la Universidad de Ottawa estudian el uso de células madre para tratar la pérdida de función muscular causada por DMD en modelos animales, con el fin de conseguir una regeneración eficiente del músculo y prevenir la pérdida progresiva de la fuerza muscular característica de la enfermedad. Se espera que los resultados de distintos estudios permitan diseñar combinaciones de estrategias eficaces para el manejo de la enfermedad.
Me gustaría saber qué es la prednisona?? Pues se menciona en este apartado que presentó beneficios en la DMD, y cuales fueron esos beneficios??
ResponderEliminarHola Patrycja, gracias por tu pregunta.
ResponderEliminarLa prednisona es un corticoesteroide. Se ha visto que en la DMD los corticoesteroides disminuyen el ritmo de deterioro de la fuerza muscular y de la función motora. Los niños con DMD pierden la capacidad de caminar a la edad media de 9,5 años si no son tratados con corticoesteroides. Los corticoesteroides se empiezan a administrar cuando el retraso del desarrollo motor comienza a notarse (entre 5-7 años). Iniciar el tratamiento en niños con DMD que ya no caminan es un asunto de decisión individual y necesita ser discutido con el médico. Algunos expertos recomiendan continuar el tratamiento una vez perdida la deambulación para preservar la fuerza de las extremidades superiores y disminuir la progresión de la escoliosis. Deben tratarse las complicaciones derivadas del uso prolongado de corticoesteroides: control del peso, administración de antagonistas H2 como protección gástrica, seguimiento y tratamiento regular de la osteoporosis y evaluación oftálmica de cataratas y glaucoma.
Incluimos más información en el apartado de tratamiento.